Leber amavrozu
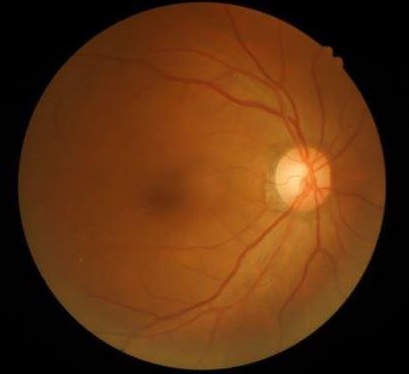 Leber amavrozu – gözün torlu qişasının işığa həssas hüceyrələrinin anadangəlmə zədələnməsi və bəzi hallarda digər ümumi pozulmalarla (böyrəklərin, MSS-nin anomaliyaları) xarakterizə olunan irsi xəstəlikdir. Bu patologiya zamanı uşağın həyatının ilk ayında və ya doğuşdan dərhal sonra nistaqm, bəbəklərin işığa reaksiyasının zəifliyi və ya ümumən olmaması meydana çıxır. Gələcəkdə uşaq gözünü ova (Françesketti simptomu), hipermetropiya (uzağı görmə) və fotofobiya (işıqdan qorxma) əmələ gələ bilər; görmənin tamamilə itirilməsi mümkündür. Diaqnostika həkim-oftalmoloqun inspeksiyası (baxış), elektroretinoqrafiya, irsi anamnezin öyrənilməsi və genetik analizin nəticələrinə əsaslanır. Leber amavrozunun spesifik müalicəsi bu günədək işlənib hazırlanmayıb.
Leber amavrozu – gözün torlu qişasının işığa həssas hüceyrələrinin anadangəlmə zədələnməsi və bəzi hallarda digər ümumi pozulmalarla (böyrəklərin, MSS-nin anomaliyaları) xarakterizə olunan irsi xəstəlikdir. Bu patologiya zamanı uşağın həyatının ilk ayında və ya doğuşdan dərhal sonra nistaqm, bəbəklərin işığa reaksiyasının zəifliyi və ya ümumən olmaması meydana çıxır. Gələcəkdə uşaq gözünü ova (Françesketti simptomu), hipermetropiya (uzağı görmə) və fotofobiya (işıqdan qorxma) əmələ gələ bilər; görmənin tamamilə itirilməsi mümkündür. Diaqnostika həkim-oftalmoloqun inspeksiyası (baxış), elektroretinoqrafiya, irsi anamnezin öyrənilməsi və genetik analizin nəticələrinə əsaslanır. Leber amavrozunun spesifik müalicəsi bu günədək işlənib hazırlanmayıb.
- Leber amavrozunun səbəbləri
- Leber amavrozunun təsnifatı
- Leber amavrozunun simptomları
- Leber amavrozunun diaqnostikası
- Leber amavrozunun müalicəsi və proqnozu
Leber amavrozu barədə ümumi məlumat
Anadangəlmə Leber amavrozu torlu qişa zülallarını, o cümlədən opsini kodlaşdıran 18 genin mutasiyasının səbəb olduğu heterogen qrup xəstəlikləri özündə cəmləyir. Amavroz ilk dəfə XIX əsrdə (1867-ci ildə) T.Leber tərəfindən təsvir olunub. O, bu xəstəliyin əsas təzahürlərini – kəfkirəbənzər nistaqm, korluq, göz dibində piqment ləkələrinin və toplantıların əmələ gəlməsini göstərmişdi. Patologiya əhali arasında orta hesabla 3: 100 000 nisbətində yayılıb. Xəstəliyin irsi ötürülməsinin əsas mexanizmi autosom-resessivdir, lakin autosom-dominant yolla ötürülən formalar da var. Leber amavrozu kişilərdə və qadınlarda eyni intensivlikdə rastlanır. Xəstəlik bütün irsi retinopatiyaların təxminən 5%-ini təşkil edir. Müasir genetika bu xəstəliyin müalicəsini işləyib hazırlayır. Leber amavrozunun bir – RPE65 geninin mutasiyası ilə əlaqəli formasında gen terapiyasının ümidləndirici nəticələri mövcuddur.
Leberin görmə sinirinin atrofiyası ayrı nozoloji vahid kimi qəbul olunur. Bu nozologiya görmə itiliyinin tədricən zəifləməsi və sonda itirilməsi ilə xaraterizə olunur. Lakin bu xəstəliyin tamamilə başqa genetik təbiəti var və o, unikal irsi ötürülməyə (ana xətti ilə) malik mitoxondrial DNT-nin zədələnməsi ilə əlaqəlidir.
Leber amavrozunun səbəbləri
 Leber amavrozunda görmənin pozulmasının əsas mexanizmi fotoreseptorların letal zədələnməsinə və məhvinə gətirib çıxaran çöpcüklər və kolbacıqlarda metabolizmin pozulmasıdır. Lakin belə dəyişikliklərin birbaşa səbəbi xəstəliyin hansı genin mutasiyası nəticəsində inkişafından asılıdır.
Leber amavrozunda görmənin pozulmasının əsas mexanizmi fotoreseptorların letal zədələnməsinə və məhvinə gətirib çıxaran çöpcüklər və kolbacıqlarda metabolizmin pozulmasıdır. Lakin belə dəyişikliklərin birbaşa səbəbi xəstəliyin hansı genin mutasiyası nəticəsində inkişafından asılıdır.
Leber amavrozunun ən geniş yayılmış tiplərindən biri (2-ci tip, LCA2) birinci xromosomda RPE65 mutant geninin mövcudluğu ilə əlaqəlidir. Bu genin 80-dən artıq mutasiyası məlumdur; bu mutasiyaların bəziləri Leber amavrozundan başqa torlu qişanın piqmentli abiotrofiyasının müəyyən formalarının yaranmasına səbəb olur. PRE65 geninin kodlaşdırdığı zülal gözün torlu qişasının piqmentli epitelində retinol metabolizminə cavabdehdir. Buna görə də genetik defekt olduqda bu proses pozulur, alternativ metabolik yollar inkişaf edir. Nəticədə fotoreseptorlarda rodopsin sintezi dayanır; bu da xəstəliyin xarakterik kliniki mənzərəsinə gətirib çıxarır. Genin mutant formaları autosom-resessiv mexanizmlə irsən ötürülür.
Leber amavrozunun daha az yayılmış forması (14-cü tip) 4-cü xromosomdakı LRAT geni ilə bağlıdır. Bu gen hepatositlərin mikrosomlarında yerləşən və gözün torlu qişasında aşkar olunmuş lesitin-retinol-asiltransferaza zülalını kodlaşdırır. Həmin ferment retinoid və vitamin A metabolizminində iştirak edir; genin mutasiyası zamanı natamam protein öz funksiyalarını tam şəkildə yerinə yetirə bilmir, nəticədə fotoreseptorların degenerasiyasının inkişafına gətirib çıxarır. Beləliklə, kliniki olaraq Leber amavrozu və ya torlu qişanın yuvenil piqmentli abiotrofiyası şəklində təzahür edir. Patologiya üçün autosom-resessiv yolla irsən ötürülmə xarakterikdir.
Leber amavrozunun 8-ci tipi tez-tez anadangəlmə korluğa gətirib çıxarır. Xəstəliyin bu formasının inkişafına 1-ci xromosomda yerləşən CRB1 genin mutasiyası cavabdehdir və autosom-resessiv yolla irsən ötürülür. Bununla yanaşı, məlum olmuşdur ki, bu genin kodlaşdırdığı zülal torlu qişanın fotoreseptorlarının və piqmentli epitelinin embrional inkişafında birbaşa iştirak edir. Leber amavrozunun bu formasının patogenezi haqda bu günədək daha dəqiq məlumatlar toplanmamışdır. 6-cı xromosomda yerləşən və Leber amavrozunun 5-ci tipinin inkişafına səbəb olan LCA5 geninin mutasiyası ilə bağlı da vəziyyət analojidir. Hazırda bu genin kodlaşdırdığı yalnız lebersilin zülalı məlumdur, lakin bu zülalın torlu qişadakı funksiyaları bəlli deyil.
Leber amavrozunun autosom-dominant yolla irsən ötürülən iki forması (CRX geninin mutasiyası ilə əlaqəli olan 7-ci tip və IMPDH1 geninin mutasiyası ilə əlaqəli 11-ci tip) da aşkar olunub. CRX geni embrional dövrdə fotoreseptorların inkişafını kontrol edən, onların böyüklərdə adekvat səviyyədə olmasını dəstəkləyən, torlu qişanın başqa proteinlərinin sintezində iştirak edən zülalı (transkripsiya faktoru hesab olunur) kodlaşdırır. Buna görə də CRX geninin mutasiyasının xarakterindən asılı olaraq Leber amavrozunun 7-ci tipinin klinikası da anadangəlmə korluqdan görmənin kifayət qədər gec və süst gedişli pisləşməsinə qədər müxtəlif olur. IMPDH1 geninin kodlaşdırdığı inozin-5′-monofosfatdehidrogenaza-1 hüceyrələrin inkişafını və nuklein turşularının sintezini tənzimləyir; lakin bu, həmin zülalın zədələnməsi zamanı Leber amavrozunun 11-ci tipinin inkişafının patogenezini aydınlaşdırmağa hələki imkan vermir.
Leber amavrozunun təsnifatı
Hazırda Leber amavrozunun 16 tipi üçün kliniki təzahürlər və müəyyən genlərin mutasiyası arasında qarşılıqlı əlaqənin mövcudluğu sübut olunub. Həmçinin belə xəstəliyin yaranmasına gətirib çıxaran daha 2 genin kəşfi ilə bağlı sübutlar da var, lakin hələ ki, bu istiqamətdə araşdırmalar davam edir.
- 1-ci tip(LCA1, ing. Leber’s congenital amaurosis) – 17-ci xromosomdakı GUCY2D geni zədələnir, autosom-resessiv yolla irsən ötürülür.
- 2-ci tip(LCA2) –1-ci xromosomdakı RPE65 geni zədələnir, autosom-resessiv yolla irsən ötürülür, bu forma gen terapiyasının müsbət nəticə verdiyi Leber amavrozunun ilk formasıdır.
- 3-cü tip(LCA3) – 14-cü xromosomdakı RDH12 geni zədələnir, autosom-resessiv yolla irsən ötürülür.
- 4-cü tip(LCA4) – 17-ci xromosomdakı AIPL1 geni zədələnir, autosom-resessiv yolla irsən ötürülür.
- 5-ci tip(LCA5) – 6-cı xromosomdakı LCA5 geni zədələnir, autosom-resessiv yolla irsən ötürülür.
- 6-cı tip(LCA6) – 14-cü xromosomdakı RPGRIP1 geni zədələnir, autosom-resessiv yolla irsən ötürülür.
- 7-ci tip(LCA7) – 19-cu xromosomdakı CRX geni zədələnir, autosom-dominant yolla irsən ötürülür. Dəyişkən kliniki mənzərə ilə xarakterizə olunur.
- 8-ci tip (LCA8) – 1-ci xromosomdakı CRB1 geni zədələnir, autosom-resessiv yolla irsən ötürülür. Statistik olaraq digər tiplərlə müqayisədə anadangəlmə korluğa daha çox səbəb olur.
- 9-cu tip (LCA9) – 1-ci xromosomdakı LCA9 geni zədələnir, autosom-resessiv yolla irsən ötürülür.
- 10-cu tip(LCA10) – 12-ci xromosomdakı CEP290 geni zədələnir, autosom-resessiv yolla irsən ötürülür.
- 11-ci tip (LCA11) – 7-ci xromosomdakı IMPDH1 geni zədələnir, autosom-dominant yolla irsən ötürülür.
- 12-ci tip (LCA12) – 1-ci xromosomdakı RD3 geni zədələnir, autosom-resessiv yolla irsən ötürülür.
- 13-cü tip (LCA13) – 14-cü xromosomdakı RDH12 geni zədələnir, autosom-resessiv yolla irsən ötürülür.
- 14-cü tip (LCA14) – 4-cü xromosomdakı LRAT geni zədələnir, autosom-resessiv yolla irsən ötürülür.
- 15-ci tip (LCA15) – 6-cı xromosomdakı TULP1 geni zədələnir, autosom-resessiv yolla irsən ötürülür.
- 16-cı tip(LCA16) – 2-ci xromosomdakı KCNJ13 geni zədələnir, autosom-resessiv yolla irsən ötürülür.
Bundan başqa, bəzən kliniki təsnifatda təkcə zədələnmiş genin adı deyil, həm də mutasiyanın xarakteri ayırd edilir. Çünki bu, Leber amavrozunun gedişinə əhəmiyyətli dərəcədə təsir edir. Bundan əlavə, eyni genin müxtəlif tip mutasiyaları tamamilə fərqli xəstəliklərə gətirib çıxara bilər; məs., CRX genində delesiyaların bəzi növləri amavroza deyil, çöpcük-kolbacıq distrofiyalarına gətirib çıxarır. RPE65, LRAT və CRB1 genlərinin bəzi mutasiyaları torlu qişanın piqmentli abiotrofiyasının müxtəlif növlərinin yaranmasına səbəb olur.
Leber amavrozunun simptomları
 Leber amavrozunun simptomatikası kifayət qədər dəyişkəndir; xəstəliyin tipindən və gen mutasiyasının xarakterindən asılıdır. Əksər hallarda uşaq doğulduqda xəstəlik müəyyənləşdirilmir, hətta göz dibinin müayinəsində dəyişikliklər kliniki halların yalnız bəzilərində müşahidə olunur. Patologiyanın inkişafından asılı olaraq valideynlər sezə bilərlər ki, uşağın baxışları ətrafdakılarda və əşyalarda ləngimir, böyük yaşlarda işığa qarşı ağrılı reaksiya verir (fotofobiya yaranır), tez-tez gözlərini ovur və onları barmaqla göstərir (Françesketti simptomu, göz-barmaq sindromu). Həyatının 2-3-cü aylarında nistaqm meydana çıxır. Bu, tez-tez Leber amavrozunun ilk təzahürlərindən biri kimi qiymətləndirilir; göz bəbəyinin işığa zəif reaksiyası və ya tamamilə olmaması aşkar olunur.
Leber amavrozunun simptomatikası kifayət qədər dəyişkəndir; xəstəliyin tipindən və gen mutasiyasının xarakterindən asılıdır. Əksər hallarda uşaq doğulduqda xəstəlik müəyyənləşdirilmir, hətta göz dibinin müayinəsində dəyişikliklər kliniki halların yalnız bəzilərində müşahidə olunur. Patologiyanın inkişafından asılı olaraq valideynlər sezə bilərlər ki, uşağın baxışları ətrafdakılarda və əşyalarda ləngimir, böyük yaşlarda işığa qarşı ağrılı reaksiya verir (fotofobiya yaranır), tez-tez gözlərini ovur və onları barmaqla göstərir (Françesketti simptomu, göz-barmaq sindromu). Həyatının 2-3-cü aylarında nistaqm meydana çıxır. Bu, tez-tez Leber amavrozunun ilk təzahürlərindən biri kimi qiymətləndirilir; göz bəbəyinin işığa zəif reaksiyası və ya tamamilə olmaması aşkar olunur.
Kliniki hallar arasında anadangəlmə korluq da müşahidə olunur. Əgər uşaq görmə funksiyasının nisbətən saxlanması ilə doğulubsa, həyatın ilk illərində verilmiş simptomlarla yanaşı onda həm də uzağı görmə (hipermetropiya), çəpgözlük, görmə itiliyinin güclü zəifləməsi inkişaf edir. Adətən, 10 yaşına qədər əksər xəstələr tamamilə kor olur. Sonralar onlarda digər oftalmoloji xəstəliklər – keratokonus, katarakta, qlaukoma də yarana bilər. Xəstəliyin bəzi tiplərində yanaşı gedən pozulmalar – MSS-nin pozulması, karlıq da müşahidə oluna bilər.
Leber amavrozunun diaqnostikası
Müasir oftalmologiyada Leber amavrozunun diaqnostikası göz dibinin müayinəsi, göz dibi dəyişikliklərinin dinamikasının monitorinqi, elektroretinoqrafiyaya əsasən aparılır. İrsi anamnezin, xəstəliyin bəzi tipləri üçün genetik sekvenləşdirmə (Genetic sequencing) ilə əsas genlərin sıralanmasının öyrənilməsi mühüm rol oynayır.
Göz dibinin müayinəsi zamanı uzun müddət (həyatın ilk bir neçə ili) heç bir dəyişiklik təyin olunmaya bilər. Amavrozun ilk, lakin qeyri-spesifik oftalmoloji simptomları nistaqm, çəpgözlük, bəbəklərin işığa reaksiyasının zəifləməsi və ya ümumiyyətlə olmamasıdır. Torlu qişada tədricən yaranan dəyişikliklər müxtəlif ölçülü, piqmentli və ya piqmentsiz ləkələr, arteriolların daralması, görmə siniri diskinin solğunlaşmasına səbəb olur. Demək olar ki, bütün xəstələrdə 8-10 yaşlarda göz dibinin periferiyasında yerləşən sümük (qara) piqmentli cisimciklər müşahidə olunur. Xarakterik əlamət torlu qişa dəyişikliklərinin görmənin funksional dəyişiklikləri ilə müqayisədə sürətlə proqressivləşməsidir; görmənin funksional pozulmaları ləng inkişaf edir. Korluğun əmələ gələnədək görmə itiliyi 0,1 və daha az təşkil edir, tez-tez uzağı görmə, fotofobiya qeyd olunur.
Yeniyetmələrdə və böyüklərdə bu simptomlara əlavə olaraq keratokonus və katarakta diaqnostika olunur. Leber amavrozunda elektroretinoqrafiya, bir qayda olaraq, bütün dalğaların amplitudunun kəskin azalmasını və ya tamamilə yoxluğunu əks etdirir. Genetik müayinələr yalnız 50-60% hallarda (daha çox yayılmış zədələnmiş genlərin tezliyi) zədələnmiş geni və mutasiyanın tipini aşkarlamağa imkan verir. Həkimlərin əksəriyyəti yalnız RPE65, CRX, CRB1, LCA5 və KCNJ13 genlərinin mutasiyasını aşkarlamaq məqsədilə sekvenləşdirmə aparırlar.
Differensial diaqnostika torlu qişanın piqmentli abiotrofiyasının müxtəlif formaları (onlarda elektroretinoqramda dalğaların amplitudu normal və ya bir qədər azalmış olaraq qalır) və görmə sinirinin atrofiyasının bəzi tipləri ilə aparılır.
Leber amavrozunun müalicəsi və proqnozu
Hazırda Leber amavrozunun istənilən tipinin spesifik müalicəsi yoxdur. Kliniki araşdırmalar mərhələsində amavrozun 2-ci tipi ilə xəstə olanların gözlərinin torlu qişasına gen mühəndisliyi üsulu ilə RPE65 geninin yeridilməsinə nail olunub; tədqiqata cəlb olunmuş pasientlərin görməsində xeyli yaxşılaşmanın əldə olunması haqda ilk məlumatlar mövcuddur. Xəstəliyin digər formalarında belə proqres hələki yoxdur. Dəstəkləyici müalicə vitaminoterapiya, damargenəldici preparatların gözdaxili inyeksiyası vasitəsilə aparılır. Hipermetropiya zamanı eynək təyin olunur.
Görmənin saxlanması zamanı proqnoz kifayət qədər xoşagəlməzdir; demək olar ki, 95% xəstədə 10 yaşına qədər görmə qabiliyyəti tamamilə itirilir. Bundan başqa, bu irsi xəstəlik MSS, böyrəklər, endokrin sistemin problemləri ilə ağırlaşa bilər. Bu səbəbdən də belə patologiyaların vaxtında aşkar olunması üçün hərtərəfli tibbi monitorinq tələb olunur.
