Villebrand xəstəliyi
 Villebrand xəstəliyi – plazma faktoru Villebrandın kəmiyyət və keyfiyyət defisiti və laxtalanma müddətinin (qanamanın) uzanması ilə təzahür edən homeostazın anadangəlmə patologiyasıdır. Xəstəlik spontan olaraq dərialtı petexiya və ekximozların əmələ gəlməsi; burun, mədə-bağırsaq traktı və uşaqlıqdan residivləşən qanaxmalar; hemartrozlar, travma və cərrahi əməliyyatlardan sonra həddən artıq qanitirmə ilə xarakterizə olunur. Diaqnoz ailə anamnezi, klinik mənzərə və homeostaz sisteminin laborator skrininqinə əsasən qoyulur. Villebrand xəstəliyi zamanı antihemofil plazma transfuziyası, yerli və ümumi hemostatik preparatlar, antifibrinolitiklər tətbiq olunur.
Villebrand xəstəliyi – plazma faktoru Villebrandın kəmiyyət və keyfiyyət defisiti və laxtalanma müddətinin (qanamanın) uzanması ilə təzahür edən homeostazın anadangəlmə patologiyasıdır. Xəstəlik spontan olaraq dərialtı petexiya və ekximozların əmələ gəlməsi; burun, mədə-bağırsaq traktı və uşaqlıqdan residivləşən qanaxmalar; hemartrozlar, travma və cərrahi əməliyyatlardan sonra həddən artıq qanitirmə ilə xarakterizə olunur. Diaqnoz ailə anamnezi, klinik mənzərə və homeostaz sisteminin laborator skrininqinə əsasən qoyulur. Villebrand xəstəliyi zamanı antihemofil plazma transfuziyası, yerli və ümumi hemostatik preparatlar, antifibrinolitiklər tətbiq olunur.
- Villebrand xəstəliyinin təsnifatı
- Villebrand xəstəliyinin səbəbləri
- Villebrand xəstəliyinin simptomları
- Villebrand xəstəliyinin diaqnostikası və müalicəsi
- Villebrand xəstəliyinin proqnozu və profilaktikası
Villebrand xəstəliyi barədə ümumi məlumat
Villebrand xəstəliyi (angiohemofiliya) – irsi hemorragik diatezlərin növüdür. Patologiya laxtalanmanın VIII – Villebrand faktorunun (VWF) olmaması və ya aktivliyinin az olması ilə əlaqədardır. Villebrand xəstəliyi geniş yayılmış koaqulyasiya patologiyasıdır; 1-2:10 000 nisbətində rastlanır. Hemorragik diatezlər arasında trombositopatiyalar və hemofiliya A-dan sonra üçüncü yeri tutur. Villebrand xəstəliyi hər iki cinsin nümayəndələrində eyni tezliklə diaqnostika olunur; lakin ağır gedişatı daha çox qadınlarda qeyd olunur. Xəstəlik birləşdirici toxuma displaziyası, bağların zəifliyi və oynaqların hipermobilliyi, yüksək dəri gərginliyi, ürək qapaqlarının prolapsı (Ehlers-Danlos sindromu) ilə müşayiət oluna bilər.
Villebrand xəstəliyinin təsnifatı
Villebrand xəstəliyinin bir neçə klinik tipləri ayırd edilir: klassik (I tip), variant formalar (II tip), ağır forma (III tip) və trombositar forma.
Xəstəliyin ən geniş yayılmış (70-80%) forması olan I tipdə plazmada Villebrand faktorunun səviyyəsi azacıq və ya mülayim azalır, bəzən normanın aşağı limitindən bir qədər az olur. Oliqomerlərin spektri dəyişilməmişdir; lakin Vinçez formasında VWF-in həddən artıq ağır multimerləri daim qeyd olunur.
II tipdə (20-30% hallarda) səviyyəsi norma daxilində olan Villebrand faktorunun aktivliyinin zəifləməsi və keyfiyyət defekti müşahidə olunur. Bunun səbəbi yüksək və orta molekulyar oliqomerlərin defisiti və ya olmaması; trombosit reseptorlarına izafi afinlik (oxşarlıq), ristomisin-kofaktor aktivliyinin azalması, VIII faktorun birləşməsinin və inaktivasiyasının pozulması ola bilər.
III tipdə plazmada Villebrand faktoru, demək olar ki, olmur, VIII faktorun aktivliyi isə aşağı olur. Trombositar tip (Villebrand psevdo-xəstəliyi) VWF-in normal miqdarında müşahidə olunur; lakin onun müvafiq dəyişilmiş trombosit reseptoru ilə birləşməsi yüksəlir.
Villebrand xəstəliyinin səbəbləri
Villebrand xəstəliyinin əsasında kompleks oliqomerlərdən (dimerlərdən multimerlərə qədər) ibarət qan plazmasının mürəkkəb qlikoproteininin – Villebrand faktorunun sintezinin kəmiyyət (I və III tip) və keyfiyyət (II tip) pozulması durur. Villebrand faktoru damar endoteli hüceyrələri və meqakariositlər tərəfindən proprotein şəklində sekresiya olunur, qana və subendotelial matriksə keçərək trombositlərin a-qranullarında və Veybl-Palad cisimciklərində (Weibel–Palade bodies) toplanır.
Villebrand faktoru damar-trombositar (birincili) və koaqulyasion (ikincili) hemostazda iştirak edir. VWF antihemofil qlobulinin (laxtalanmanın VIII faktoru) subvahididir; onun stabilliyini və vaxtından əvvəl inaktivasiyasından müdafiəni təmin edir. Spesifik reseptorların mövcudluğu səbəbindən Villebrand faktoru qan lövhəciklərinin (trombositlərin) subepitelial strukturlara qüsurlu adheziyasına və qan damarlarının zədələndiyi nahiyələrdə bir-birləri ilə aqreqasiyasına revac verir.
Normada plazmada VWF-in səviyyəsi 10 mq/l-dir; fiziki aktivlik, hamiləlik, stress, iltihabi-infeksion proseslər, estrogen qəbulu zamanı müvəqqəti yüksəlir; l qan qrupu olan insanlarda konstitusional olaraq az olur. Villebrand faktorunun aktivliyi onun molekulyar kütləsindən asılıdır; nisbətən yüksək trombogen potensial ən iri multimerlərdə qeyd olunur.
Villebrand xəstəliyi genetik patologiyadır; 12-ci xromosomda yerləşən VWF faktorunun geninin mutasiyası nəticəsində inkişaf edir. Xəstəliyin I və II tipi autosom-dominant tip üzrə natamam penetrantlıqla (xəstələr heteroziqotdur), III tipi autosom-resessiv tip üzrə (xəstələr homoziqotdur) ötürülür. Villebrand xəstəliyinin III tipində VWF geninin böyük hissəsinin delesiyası, mutasiyası və ya bu defektlərin kombinasiyası rol oynayır. Bu zaman hər iki valideyn, adətən, xəstəliyin I tipinin yüngül gedişatına malik olur.
Villebrand xəstəliyinin qazanılmış formaları sistem (sistemli qırmızı qurdeşənəyi, revmatoid artrit), ürək (aortal qapağın stenozu), onkoloji (nefroblastoma, Vilms şişi, makroqlobulinemiya) xəstəliklər fonunda çoxsaylı hemotransfuziyalardan sonra ağırlaşma kimi meydana çıxa bilər. Xəstəliyin bu formaları VWF-ə qarşı autoantitellərin əmələ gəlməsi, şiş hüceyrələri tərəfindən oliqomerlərin seçici absorbsiyası və ya trombositlərin membranlarının defekti ilə əlaqəlidir.
Villebrand xəstəliyinin simptomları
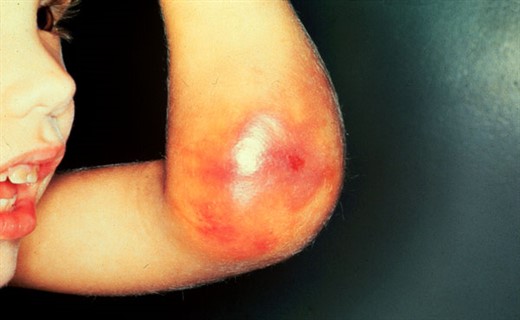
Villebrand xəstəliyi müxtəlif intensivlikli hemorragik sindromla təzahür edir. Xəstəliyin ağırlığından və variantından asılı olaraq əsasən petexial-göyümtül ləkələr, göyümtül ləkələr-hematom, nadir hallarda hematom tipli olur.
Villebrand xəstəliyinin I və II tipinin yüngül formaları spontan burun qanaxmaları, dəri içinə və dərialtı az və mülayim qansızma (petexiya, ekximoz), travmalardan (kiçik və dərin olmayan kəsilmiş yaralar) və cərrahi manipulyasiyalardan (diş ekstraksiyası, tonzillektomiya və s.) sonra davamlı qanaxma ilə xarakterizə olunur. Qızlarda menorragiya, uşaqlıq qanaxmaları, uşaq dünyaya gətirən qadınlarda doğuş zamanı həddən artıq qan itkisi qeyd olunur.
Villebrand xəstəliyinin I və II tipinin ağır formalarında və III tipində klinik mənzərə hemofiliya simptomlarını xatırlada bilər. Tez-tez dərialtı hemorragiyalar, yumşaq toxumaların ağrılı hematomaları, inyeksiya yerindən qanaxma meydana çıxır. İri oynaqlara qansızma (hemartrozlar), cərrahi əməliyyatlar və travmalar zamanı uzun müddət dəf edilə bilməyən qanaxma; burundan, diş ətindən, mədə-bağırsaq traktından və sidik yollarından güclü qanaxma baş verir. Kobud posttravmatik çapıqların formalaşması xarakterikdir. Villebrand xəstəliyinin ağır gedişində hemorragik sindrom uşağın həyatının ilk aylarından özünü göstərir. Patologiya zamanı hemosindrom fəsadlaşma və təzahürlərin, demək olar ki, və ya ümumiyyətlə yox olması ilə növbələşir. Lakin xəstəliyin ağır gedişli formaları ağır posthemorragik anemiyaya gətirib çıxara bilər.
Villebrand xəstəliyinin diaqnostikası və müalicəsi
Villebrand xəstəliyinin diaqnostikasında ailə anamnezi, klinik mənzərə, plazma hemostazı və damar-trombositar laborator skrininq mühüm rol oynayır. Qanın ümumi və biokimyəvi analizi; laxtalanma müddəti, trombosit və fibrinogen səviyyəsinin müəyyənləşdirilməsi ilə koaquloqramma; protrombin indeksi (PTİ) və aktivləşmiş hissəvi tromboplastin müddəti (AHTM), çimdik və juqut sınaqları aparılır. Ümumi müayinələrdən qan qrupunun müəyyənləşdirilməsi, sidiyin ümumi analizi, nəcisin gizli qana görə analizi, abdominal USM tövsiyə olunur.
Villebrand xəstəliyinin təsdiqi üçün qan zərdabında VWF-in səviyyəsi və aktivliyi, immunoelektroforez və İFA metodlarının tətbiqi ilə ristosetin-kofaktor aktivliyi müəyyən olunur. Xəstəliyin II tipində, VWF və VIII faktorun səviyyəsi normal olduqda trombositlərin aktivasiya faktoru (PAF – platelet-activating factor), laxtalanmanın VIII faktorunun aktivliyi, trobositlərin aqreqasiyasını müəyyənləşdirmək informativdir. Villebrand xəstəliyindən əziyyət çəkən pasiyentlər üçün qan zərdabında VWF-in aktivliyinin və səviyyəsinin azalması, qanaxmanın müddətinin və aktivləşmiş hissəvi tromboplastin müddətinin uzanması, trombositlərin adheziya və aqreqasiya funksiyalarının pozulması xarakterikdir.
Villebrand xəstəliyi hemofiliya, irsi trombositopatiyalarla differensial diaqnostika olunmalıdır. Hematoloq və genetik konsultasiyasından başqa otolarinqoloq, stomatoloq, ginekoloq və qastroenteroloq müayinəsi də aparılır.
Az simptomlu və mülayim təzahür edən hemosindromlu Villebrand xəstəliyinin müntəzəm müalicəsi aparılmır; lakin pasiyentlərdə qanaxma riski yüksək olaraq qalır. Müalicə doğuş, travmalar, menorragiya, hemartoz zamanı qanaxma yarandıqda, cərrahi və stomatoloji müdaxilələrdən əvvəl profilaktik məqsədlə təyin olunur. Müalicənin məqsədi defisit laxtalanma faktorlarının lazımi minimal səviyyəsini təmin etməkdən ibarətdir.
Əvəzedici terapiya kimi hemofiliya ilə müqayisədə az dozalarda antihemofil plazma və kriopresipitat (VWF-in yüksək miqdarı ilə) göstərişdir. I tip Villebrand xəstəliyi zamanı qanaxmanı dayandırmaq məqsədilə desmopressinin təyini effektlidir. Hemoragiyanın yüngül və orta-ağırlıqlı formalarında aminokapron turşusu, traneksam turşusu təyin oluna bilər. Yaraların qanamasının dayandırılması məqsədilə hemostatik süngər, fibrin yapışqanı istifadə olunur. Təkrarlanan uşaqlıq qanaxmalarında KOK (kombinə olunmuş oral kontraseptivlər) təyin olunur; müsbət effekt olmadıqda histerektomiya (uşaqlığın cərrahi kəsilməsi) icra olunur.
Villebrand xəstəliyinin proqnoz və profilaktikası
Villebrand xəstəliyinin adekvat hemostatik müalicəsi aparıldıqda xəstəlik, adətən, xoşxassəli gedişata malik olur. Patologiyanın ağır gedişi güclü posthemorragik anemiyaya; doğuşdan, ciddi travmalardan və cərrahi əməliyyatlardan sonra fatal qanaxmaya, bəzən subaraxnoidal qansızmaya və hemorragik insulta gətirib çıxara bilər. Villebrand xəstəliyinin profilaktikası məqsədilə xəstələr (o cümlədən qohumlar) arasında nigahın istisnası, diaqnoz qoyulduqda QSİƏP, antiaqreqantlar qəbul etməmək, travmalardan qaçmaq, həkimin tövsiyələrinə dəqiq əməl etməkdən ibarətdir.
